
任何MCMC方案的目标都是从“目标”分布产生样本。
在这种情况下,我们将使用平均值为1的指数分布作为我们的目标分布。所以我们从定义目标密度开始:
target = function(x){
if(x<0){
return(0)}
else {
return( exp(-x))
}
}可下载资源
定义了函数之后,我们现在可以用它来计算几个值(只是为了说明函数的概念):
target(1)[1] 0.3678794target(-1)[1] 0接下来,我们将规划一个Metropolis-Hastings方案,从与目标成比例的分布中进行抽样
x[1] = 3 #这只是一个起始值,我设置为3
for(i in 2:1000){
A = target(proposedx)/target(currentx)
if(runif(1)<A){
x[i] = proposedx # 接受概率min(1,a)
} else {
x[i] = currentx #否则“拒绝”行动,保持原样
} 注意,x是马尔可夫链的实现。我们可以画几个x的图:

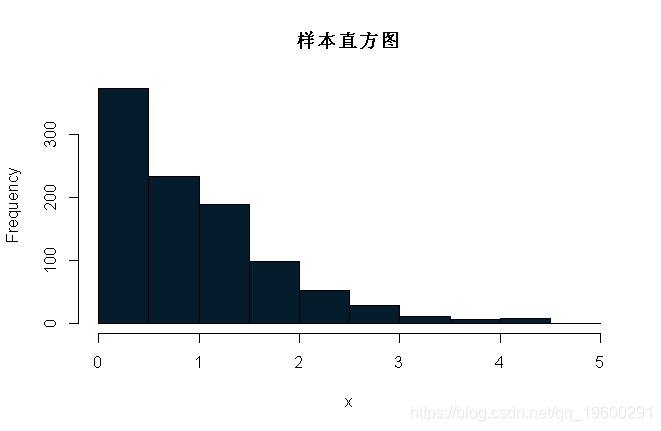
我们可以将其封装在一个mcmc函数中,以使代码更整洁,这样更改起始值和提议分布更容易
for(i in 2:niter){
currentx = x[i-1]
proposedx = rnorm(1,mean=currentx,sd=proposalsd)
A = target(proposedx)/target(currentx)
if(runif(1)<A){
x[i] = proposedx # 接受概率min(1,a)
} else {
x[i] = currentx # 否则“拒绝”行动,保持原样
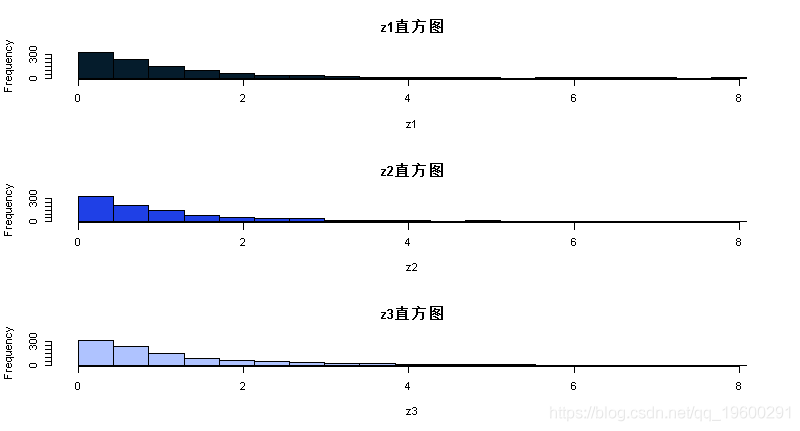
} 现在我们将运行MCMC方案3次,看看结果有多相似:
z1=MCMC(1000,3,1)
z2=MCMC(1000,3,1)
z3=MCMC(1000,3,1)
plot(z1,type="l") 
par(mfcol=c(3,1)) #告诉R将3个图形放在一个页面上
hist(z1,breaks=seq(0,maxz,length=20)) 
练习
使用函数easyMCMC了解以下内容:
- 不同的起始值如何影响MCMC方案?
- 较大/较小的提案标准差有什么影响?
- 尝试将目标函数更改为
随时关注您喜欢的主题
target = function(x){
return((x>0 & x <1) + (x>2 & x<3))
}这个目标看起来像什么?如果提议sd太小怎么办?(例如,尝试1和0.1)
例2:估计等位基因频率
在对双等位基因座的基因型(如具有AA和AA等位基因的基因座)进行建模时,一个标准的假设是群体是“随机”的。这意味着如果p是等位基因AA的频率,那么基因型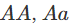
和
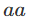
将分别具有频率
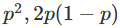
和
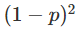
。
p一个简单的先验是假设它在[0,1]上是均匀的。 假设我们抽样n个个体,观察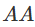
基因型
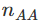
、
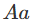
基因型
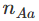
和
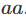
基因型
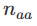
。
下面的R代码给出了一个简短的MCMC例程,可以从p的后验分布中进行采样。请尝试遍历该代码,看看它是如何工作的。
prior = function(p){
if((p<0) || (p>1)){ # || 这里意思是“或”
return(0)}
else{
return(1)}
}
likelihood = function(p, nAA, nAa, naa){
return(p^(2*nAA) * (2*p*(1-p))^nAa * (1-p)^(2*naa))
}
psampler = function(nAA, nAa, naa){
for(i in 2:niter){
if(runif(1)<A){
p[i] = newp # 接受概率min(1,a)
} else {
p[i] = currentp # 否则“拒绝”行动,保持原样
} 对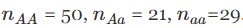
运行此样本。
现在用一些R代码来比较后验样本和理论后验样本(在这种情况下可以通过分析获得;因为我们观察到121个As和79个as,在200个样本中,p的后验样本是β(121+1,79+1)。
hist(z,prob=T)
lines(x,dbeta(x,122, 80)) # 在直方图上叠加β密度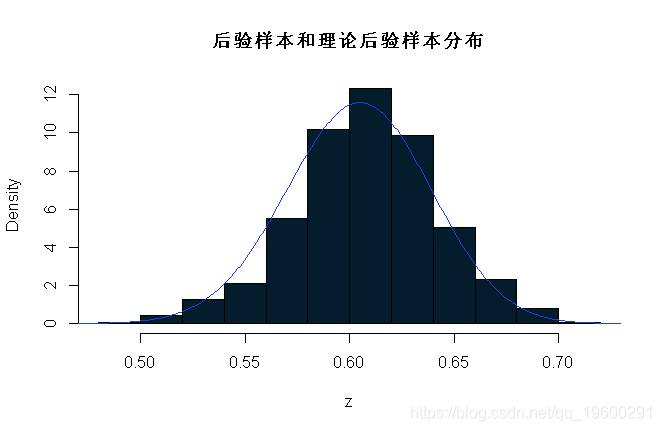
您也可能希望将前5000 z的值丢弃为“burnin”(预烧期)。这里有一种方法,在R中仅选择最后5000 z
hist(z[5001:10000])
练习
研究起点和提案标准偏差如何影响算法的收敛性。
例3:估计等位基因频率和近交系数
在大多数情况下,将f作为种群特征来对待是很自然的,因此假设f在各个位点上是恒定的。
一个复杂一点的替代方法是假设人们有一种倾向,即人们会与比“随机”关系更密切的其他人近交(例如,在地理结构上的人口中可能会发生这种情况)。一个简单的方法是引入一个额外的参数,即“近亲繁殖系数”f,并假设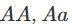
和
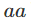
基因型有频率
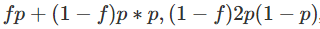
和
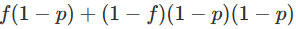
。
请注意,f和p都被约束为介于0和1之间(包括0和1)。对于这两个参数中的每一个,一个简单的先验是假设它们在[0,1]上是独立的。 假设我们抽样n个个体,观察基因型
、
基因型
和
基因型
。
练习:
- 编写一个短的MCMC程序,从f和p的联合分布中取样。
sampler = function(){
f[1] = fstartval
p[1] = pstartval
for(i in 2:niter){
currentf = f[i-1]
currentp = p[i-1]
newf = currentf +
newp = currentp +
}
return(list(f=f,p=p)) # 返回一个包含两个名为f和p的元素的“list”
}- 使用此样本获得f和p的点估计(例如,使用后验平均数)和f和p的区间估计(例如,90%后验置信区间),数据:
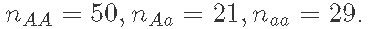
附录:GIBBS采样
您也可以用Gibbs采样器解决这个问题
为此,您将想要使用以下“潜在变量”表示模型:
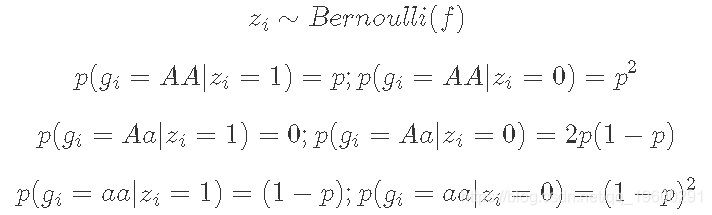
将zi相加得到与上述相同的模型:
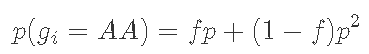
 Python Agent多GPU随机变分推断SVI加速层次贝叶斯价格弹性估计|附智能体代码数据
Python Agent多GPU随机变分推断SVI加速层次贝叶斯价格弹性估计|附智能体代码数据 Python用AI对零售商品层次贝叶斯模型价格弹性估计与个性化定价|附AI智能体、代码和数据
Python用AI对零售商品层次贝叶斯模型价格弹性估计与个性化定价|附AI智能体、代码和数据 专题:Python实现贝叶斯线性回归与MCMC采样数据可视化分析2实例|附代码数据
专题:Python实现贝叶斯线性回归与MCMC采样数据可视化分析2实例|附代码数据

