
与普通的扩散研究不同,网络扩散开始考虑网络结构对于扩散过程的影响。 这里介绍一个使用R模拟网络扩散的例子。
基本的算法非常简单: 生成一个网络:g(V, E)。 随机选择一个或几个节点作为种子(seeds)。 每个感染者以概率p(可视作该节点的传染能力,通常表示为β)影响与其相连的节点。
可下载资源
其实这是一个最简单的SI模型在网络中的实现。S表示可感染(susceptible), I表示被感染(infected)。易感态-感染态-恢复态(SIR)模型用以描述水痘和麻疹这类患者能完全康复并获得终身免疫力的流行病。
SIR模型定义
SIR模型是一种传播模型,是信息传播过程的抽象描述。
SIR模型是传染病模型中最经典的模型,其中S表示易感者,I表示感染者,R表示移除者。
S:Susceptible,易感者
I:Infective,感染者
R:Removal,移除者
SIR模型的应用
SIR模型应用于信息传播的研究。
传播过程大致如下:最初,所有的节点都处于易感染状态。然后,部分节点接触到信息后,变成感染状态,这些感染状态的节点试着去感染其他易感染状态的节点,或者进入恢复状态。感染一个节点即传递信息或者对某事的态度。恢复状态,即免疫,处于恢复状态的节点不再参与信息的传播。
SIR的微分方程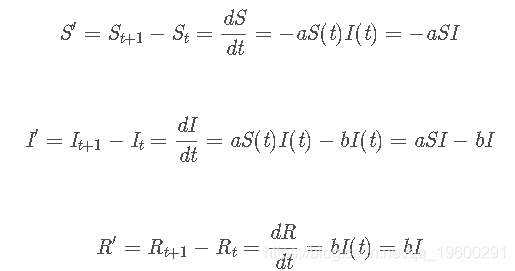
a为感染率、b恢复率
注意:
t为某个时刻,例如t=1,S(1)为第一天易感人群的人数。
无论t为什么时刻,总人数是不变的,即N(t)=S(t)+I(t)+R(t)。
人口总数总保持一个常数,即N(t)=K,不考虑人口的出生、死亡、迁移等因素。
对于SIR流行病传播模型,任意时刻节点只能处于易感态(S)或感染态(I)或恢复态(R)。
易感态节点表示未被流行病感染的个体,且可能被感染;感染态节点表示已经被流行病感染且具有传播能力;恢复态节点则表示曾感染流行病且完全康复。与SIS模型类似,每一时间步内,每个感染态节点以概率λ 尝试感染它的邻居易感态节点,并以概率γ 变为恢复态。
S = S(t)是易感个体的数量, I = I(t)是被感染的个体的数目, R = R(t)是恢复的个体的数目。
第二组因变量代表在三个类别的总人口的比例。所以,如果N是总人口(790万在我们的例子),我们有
S(T)= S(T)/ N,人口的易感部分, Ⅰ(T)= I(t)的/ N的人口感染分数并 R(T)= R(t)的/ N,人口的康复部分。
解这个微分方程,我们可以得到累计增长曲线的表达式。有趣的是,这是一个logistic增长,具有明显的S型曲线(S-shaped curve)特征。该模型在初期跨越临界点之后增长较快,后期则变得缓慢。 因而可以用来描述和拟合创新扩散过程(diffusion of innovations)。 当然,对疾病传播而言,SI模型是非常初级的(naive),主要因为受感染的个体以一定的概率恢复健康,或者继续进入可以被感染状态(S,据此扩展为SIS模型)或者转为免疫状态(R,据此扩展为SIR模型)。
免疫表示为R,用γγ代表免疫概率(removal or recovery rate)。对于信息扩散而言,这种考虑暂时是不需要的。
第一步,生成网络。
规则网
g =graph.tree(size, children =2); plot(g)
免疫表示为R,用γγ代表免疫概率(removal or recovery rate)。对于信息扩散而言,这种考虑暂时是不需要的。
第一步,生成网络。
规则网
g =graph.tree(size, children =2); plot(g)
g =graph.star(size); plot(g)
g =graph.full(size); plot(g)
g =graph.ring(size); plot(g)
g =connect.neighborhood(graph.ring(size), 2); plot(g) # 最近邻耦合网络
# 随机网络
g =erdos.renyi.game(size, 0.1)# 小世界网络
g = rewire.edges(erdos.renyi.game(size, 0.1), prob = 0.8 )# 无标度网络
g =barabasi.game(size) ; plot(g)第二步,随机选取一个或n个随机种子。
# initiate the diffusers
seeds_num =1 diffusers =sample(V(g),seeds_num) ;
diffusers
## + 1/50 vertex:
## [1] 43
infected =list()
infected[[1]]=diffusers# 第三步,传染能力
在这个简单的例子中,每个节点的传染能力是0.5,即与其相连的节点以0.5的概率被其感染,每个节点的回复能力是0.5,即其以0.5的概率被其回复。在R中的实现是通过抛硬币的方式来实现的。
## [1] 0
显然,这很容易扩展到更一般的情况,比如节点的平均感染能力是0.128,那么可以这么写: 节点的平均回复能力是0.1,那么可以这么写
p =0.128
coins =c(rep(1, p*1000), rep(0,(1-p)*1000))
sample(coins, 1, replace=TRUE, prob=rep(1/n, n))
## [1] 0
n =length(coins2)
sample(coins2, 1, replace=TRUE, prob=rep(1/n, n))
## [1] 0当然最重要的一步是要能按照“时间”更新网络节点被感染的信息。
keep =unlist(lapply(nearest_neighbors[,2], toss))
new_infected =as.numeric(as.character(nearest_neighbors[,1][keep >=1]))
diffusers =unique(c(as.numeric(diffusers), new_infected))
return(diffusers)}
set.seed(1);开启扩散过程!
先看看S曲线吧:
# # "growth_curve"num_cum =unlist(lapply(1:i, function(x) length(infected[[x]]) ))
p_cum =num_cum time =1:i
## Large initial population size (X=1000)
parms <-c(beta=0.01, gamma=0.1)
x0 <-c(S=49,I=1,R=0)a <-c("beta*S*I","gamma*I")
nu <-matrix(c(-1,0,+1,-1,0,+1),nrow=3,byrow=TRUE)
out <-ssa(x0,a,nu,parms,tf=4,simName="SIR model")为了可视化这个扩散的过程,我们用红色来标记被感染者。
# generate a palette#
plot(g, layout =layout.old)
set.seed(1)#
library(animation)# start the plot
m =1same=numeric(0)
for(m in 2:length(health))
if(length(setdiff(health[[m ]],health[[m -1 ]]) )==0){same=c(same,m)
}
health=health[-same]
infected=infected[-same]# 如同在Netlogo里一样,我们可以把网络扩散与增长曲线同时展示出来:
set.seed(1)
# start the plot
m =1
p_cum=numeric(0)
h_cum=numeric(0)
i_cum=numeric(0)
while( m<50 ) {# start the plot
layout(matrix(c(1, 2, 1, 3), 2,2, byrow =TRUE), widths=c(3,1), heights=c(1, 1))
V(g)$color = "white"
V(g)$color[V(g)%in%infected[[m ]] ] = "red"
V(g)$color[V(g)%in%health[[m ]]] = "green"
if(m<=length(infected))
plot(pp~time, type ="h", ylab ="PDF", xlab ="Time",xlim =c(0,i), ylim =c(0,1), frame.plot =FALSE)
m =m +1
}可下载资源
关于作者
Kaizong Ye是拓端研究室(TRL)的研究员。在此对他对本文所作的贡献表示诚挚感谢,他在上海财经大学完成了统计学专业的硕士学位,专注人工智能领域。擅长Python.Matlab仿真、视觉处理、神经网络、数据分析。
本文借鉴了作者最近为《R语言数据分析挖掘必知必会 》课堂做的准备。
非常感谢您阅读本文,如需帮助请联系我们!
 Python扩散模型GAN无监督行人重识别数据增强性能对比研究|附数据代码
Python扩散模型GAN无监督行人重识别数据增强性能对比研究|附数据代码 LSTM-Transformer混合模型与多源时空数据的全球水平面辐照度预测:Python实现、模型对比与消融分析 |附代码与数据
LSTM-Transformer混合模型与多源时空数据的全球水平面辐照度预测:Python实现、模型对比与消融分析 |附代码与数据 Python用Seedream4.5图像生成模型API调用与多场景应用|附代码教程
Python用Seedream4.5图像生成模型API调用与多场景应用|附代码教程 R语言广义加性模型GAM、Tweedie分布的SaaS客户生命周期价值CLV预测研究——非线性关系捕捉与异方差性适配创新|附代码数据
R语言广义加性模型GAM、Tweedie分布的SaaS客户生命周期价值CLV预测研究——非线性关系捕捉与异方差性适配创新|附代码数据



